PrecisionFDA
Consistency Challenge
Engage and improve DNA test results with our first community challenge
- Rafael Aldana
- Hanying Feng
- Brendan Gallagher
- Jun Ye
- Rafael Aldana
- Hanying Feng
- Brendan Gallagher
- Jun Ye
- Deepak Grover
We received a total of 21 entries to the challenge, summarized in the following table. The entries are sorted in order of date of submission.
We would like to acknowledge and thank all of those who participated in the precisionFDA Consistency Challenge, for their engagement and contributions. We hope that everyone will feel like a winner. After considering the performance in reproducibility and accuracy comparisons, as well as other parameters, we have decided to hand out awards and recognitions, as illustrated in the table.
| Label | Submitter | Organization | Awards | Reproducibility recognitions | Accuracy recognitions | Other recognitions |
|||
|---|---|---|---|---|---|---|---|---|---|
| Determinism | Concordance | Recall | Precision | F-measure | |||||
| ebanks-nist* | Eric Banks | ||||||||
| raldana-sentieon | Rafael Aldana et al. | Sentieon |
Performance
Reproducibility |
deterministic | Highest | High | High | High | |
| dgrover-gatk | Deepak Grover | Sanofi-Genzyme | Accuracy | High | Highest | Highest | Highest | extra-credit | |
| jedwards-sentieon | Jeremy Edwards | UNM | deterministic | High | High | High | High | ||
| ckim-gatk | Changhoon Kim | Macrogen | High | ||||||
| ckim-vqsr | Changhoon Kim | Macrogen | deterministic | High | |||||
| ckim-isaac | Changhoon Kim | Macrogen | deterministic | High | High | ||||
| ckim-dragen | Changhoon Kim | Macrogen | High | High | High | High | |||
| ckim-genalice | Changhoon Kim | Macrogen | |||||||
| mlin-fermikit | Mike Lin | DNAnexus Science | pfda-apps | ||||||
| ciseli-custom | Christian Iseli et al. | SIB | deterministic | ||||||
| astatham-gatk | Aaron Statham et al. | KCCG | High | High | |||||
| anovak-vg | Adam Novak et al. | vgteam | heroic-effort | ||||||
| mmohiyuddin-mixed | Marghoob Mohiyuddin et al. | Roche | High | High | High | High | |||
| cchapple-mixed | Charles Chapple et al. | Saphetor | High | High | High | ||||
| amark-mixed | Adam Mark | Avera | High | High | High | ||||
| jharris-gatk | Jason Harris | Personalis | deterministic | High | High | ||||
| asubramanian-gatk | Ayshwarya Subramanian et al. | Broad Institute | High | High | High | ||||
| egarrison-hhga | Erik Garrison et al. | deterministic | High | High | |||||
| xli-custom | Xiang Li et al. | Pathway Genomics | deterministic | ||||||
| rlopez-custom** | Rene Lopez et al. | MyBioinformatician | promising | ||||||
| Label | VCF Files | Comparisons |
|---|---|---|
| ebanks-nist* | G1, G2, H | R1, R2, A1a, A1b, A2 |
| raldana-sentieon | G1, G2, H | R1, R2, A1a, A1b, A2 |
| dgrover-gatk | G1, G2, H | R1, R2, A1a, A1b, A2 |
| jedwards-sentieon | G1, G2, H | R1, R2, A1a, A1b, A2 |
| ckim-gatk | G1, G2, H | R1, R2, A1a, A1b, A2 |
| ckim-vqsr | G1, G2, H | R1, R2, A1a, A1b, A2 |
| ckim-isaac | G1, G2, H | R1, R2, A1a, A1b, A2 |
| ckim-dragen | G1, G2, H | R1, R2, A1a, A1b, A2 |
| ckim-genalice | G1, G2, H | R1, R2, A1a, A1b, A2 |
| mlin-fermikit | G1, G2, H | R1, R2, A1a, A1b, A2 |
| ciseli-custom | G1, G2, H | R1, R2, A1a, A1b, A2 |
| astatham-gatk | G1, G2, H | R1, R2, A1a, A1b, A2 |
| anovak-vg | G1, G2, H | R1, R2, A1a, A1b, A2 |
| mmohiyuddin-mixed | G1, G2, H | R1, R2, A1a, A1b, A2 |
| cchapple-mixed | G1, G2, H | R1, R2, A1a, A1b, A2 |
| amark-mixed | G1, G2, H | R1, R2, A1a, A1b, A2 |
| jharris-gatk | G1, G2, H | R1, R2, A1a, A1b, A2 |
| asubramanian-gatk | G1, G2, H | R1, R2, A1a, A1b, A2 |
| egarrison-hhga | G1, G2, H | R1, R2, A1a, A1b, A2 |
| xli-custom | G1, G2, H | R1, R2, A1a, A1b, A2 |
| rlopez-custom** | G1, G2, H | R1, R2, A1a, A1b, A2 |
*ebanks-nist: This entry was not considered because the entry submitted the NIST benchmark set as the answer
**rlopez-custom: This entry was not considered because the VCF files were not submitted within the challenge timeframe
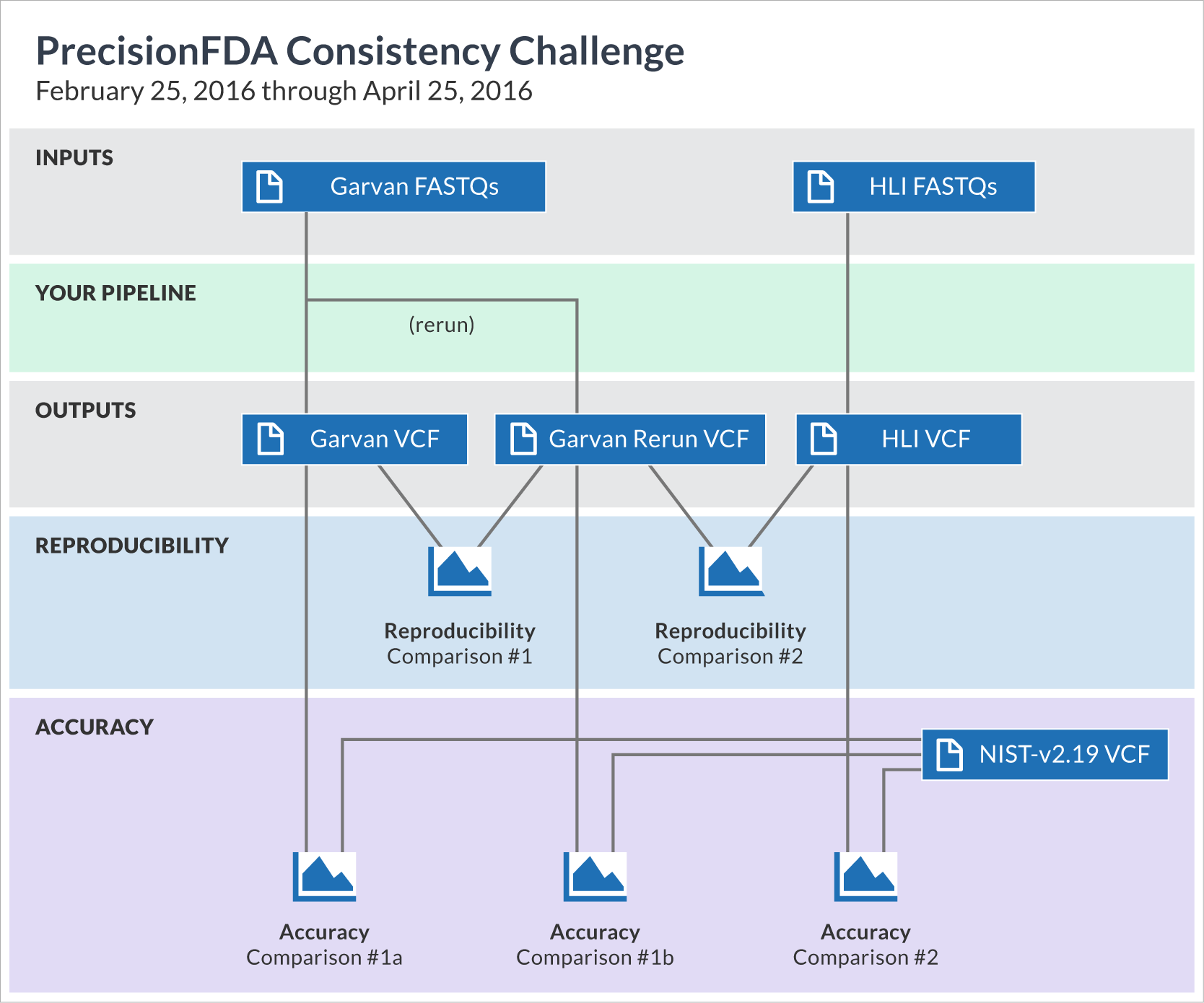
To aid in the presentation of results, we decided to give each entry a unique label, comprised of the name of the submitting user as well as a short mnemonic keyword representing the pipeline. (These keywords are merely indicative of each pipeline's main component, hence somewhat subjective; for a more faithful description of each pipeline, refer to the full text that accompanied each submission by following the label links). Each entry consists of three submitted VCF files (Garvan, Garvan-rerun, and HLI) and five comparisons (reproducibility 1 & 2; accuracy 1a, 1b & 2), as shown in the "Datasets" tab. The entries are sorted in order of date of submission.
We are handing out the following community challenge awards:
- overall-performance to the entry submitted by Rafael Aldana et al. from Sentieon, for their overall high performance in both reproducibility and accuracy.
- reproducibility to the entry submitted by Rafael Aldana et al. from Sentieon, for achieving the highest concordance and determinism.
- accuracy to the entry submitted by Deepak Grover from Sanofi-Genzyme, for achieving the highest accuracy.
We are also recognizing particular entries based on the following:
- reproducibility across the same input ( deterministic )
- reproducibility across different inputs ( highest-concordance , high-concordance )
- accuracy ( highest-recall , high-recall , highest-precision , high-precision , highest-f-measure , high-f-measure )
- miscellaneous criteria ( extra-credit , pfda-apps , heroic-effort , promising )
For more information about the determination of these recognitions, refer to the respective sections of this web page.
If the exact same pipeline runs twice on the exact same input, does it produce the same output? The reproducibility comparison 1 investigates exactly that, by comparing the Garvan VCF to the Garvan Rerun VCF (which are the results of running the same pipeline on the same sequencing data). In this comparison, true positives represent the common variants, whereas false positives and false negatives represent the unique variants in each file. The following table summarizes the performance of each entry with respect to number of common and unique variants in the reproducibility comparison 1. The entries are sorted by number of unique variants; since all the deterministic entries have no unique variants, those are sorted by submission date.
| Label | Repro.1 | Common (TP) | Unique (FP+FN) | Recognition |
|---|---|---|---|---|
| raldana-sentieon | Link | 4,600,753 | 0 | deterministic |
| jedwards-sentieon | Link | 4,662,359 | 0 | deterministic |
| ckim-vqsr | Link | 4,144,162 | 0 | deterministic |
| ckim-isaac | Link | 4,132,671 | 0 | deterministic |
| ciseli-custom | Link | 3,251,498 | 0 | deterministic |
| jharris-gatk | Link | 4,359,277 | 0 | deterministic |
| egarrison-hhga | Link | 3,131,589 | 0 | deterministic |
| xli-custom | Link | 4,984,488 | 0 | deterministic |
| cchapple-mixed | Link | 4,629,987 | 671 | |
| anovak-vg | Link | 4,988,670 | 851 | |
| ckim-gatk | Link | 4,689,121 | 2,293 | |
| ckim-dragen | Link | 4,720,819 | 6,342 | |
| ckim-genalice | Link | 4,611,442 | 7,295 | |
| dgrover-gatk | Link | 4,612,455 | 12,645 | |
| mlin-fermikit | Link | 4,536,436 | 13,105 | |
| astatham-gatk | Link | 4,532,335 | 24,120 | |
| amark-mixed | Link | 3,129,181 | 62,880 | |
| asubramanian-gatk | Link | 4,378,027 | 114,285 | |
| ebanks-nist | Link | 3,151,019 | 0 | [#1] |
| mmohiyuddin-mixed | Link | 4,632,204 | 0 | [#2] |
| rlopez-custom | Link | 3,139,985 | 0 | [#3] |
[#1] This entry was not considered because the entry submitted the NIST benchmark set as the answer
[#2] This entry was not considered because the Garvan rerun VCF file was not submitted within the challenge timeframe
[#3] This entry was not considered because the VCF files were not submitted within the challenge timeframe
We would like to recognize all entries which exhibited deterministic behavior (zero unique variants). These entries produced identical results across invocations. The remaining entries showed some degree of difference, ranging from a small amount of unique variants all the way to a hundred thousand unique variants. As suggested by some of the participants, the lack of determinism may in part be attributed to parallelization and a few other factors. One participant said: "We think the multi-threading option used during our alignment step can lead to non-deterministic answers and hence, slight variations in results. It would be useful to note how the different entries vary in this result and which factors or differences the community decides really matter in the day-to-day running of a clinical genomics facility." Similarly, another participant said: "Our pipeline, by design, must be 100% repeatable between runs, i.e., have zero run-to-run difference. This can be achieved by rigorous software design: no random numbers, no downsampling, and rigorous parallelization hence zero dependency on the number of threads."
Another question of interest is how reproducible is a result on the same biological sample and instrument but across different sites and library preps. The reproducibility comparison 2 quantifies the amount of similarity between results obtained using the Garvan and HLI datasets respectively. These datasets were generated from Coriell NA12878 DNA on a HiSeq X Ten instrument, but with different versions of the TruSeq Nano DNA Library Prep kit (v2.5 for Garvan; and v1 for HLI), and at different sequencing sites. Just like with reproducibility comparison 1, true positives represent the common variants, whereas false positives and false negatives represent the unique variants. (In all comparisons, it should be noted that if a variant is found in both the sets being compared but with different zygosity, then it is counted as both a false positive and a false negative. This is because the comparison operates at the level of genotypes and not at the level of alleles). We appreciated that a single metric, concordance, defined as (common)/(common+unique) captures the essence of this comparison: it rewards not only a decrease in the number of unique variants, but also an increase in reproducible variants. The following table shows this concordance metric for the reproducibility comparison 2. The entries are sorted by decreasing concordance.
| Label | Repro.2 | Common (TP) | Unique (FP+FN) | Concordance | Recognition |
|---|---|---|---|---|---|
| raldana-sentieon | Link | 4,305,175 | 402,872 | 91.44% | Highest |
| ckim-isaac | Link | 3,865,697 | 379,207 | 91.07% | High |
| jedwards-sentieon | Link | 4,346,524 | 450,490 | 90.61% | High |
| cchapple-mixed | Link | 4,309,008 | 451,004 | 90.53% | High |
| ckim-dragen | Link | 4,408,487 | 476,797 | 90.24% | High |
| dgrover-gatk | Link | 4,320,164 | 469,875 | 90.19% | High |
| mmohiyuddin-mixed | Link | 4,305,204 | 471,741 | 90.12% | High |
| asubramanian-gatk | Link | 4,118,928 | 457,085 | 90.01% | High |
| astatham-gatk | Link | 4,218,797 | 489,112 | 89.61% | |
| jharris-gatk | Link | 4,054,823 | 477,592 | 89.46% | |
| xli-custom | Link | 4,586,333 | 617,194 | 88.14% | |
| ckim-gatk | Link | 4,194,953 | 650,105 | 86.58% | |
| ckim-genalice | Link | 4,181,476 | 719,864 | 85.31% | |
| mlin-fermikit | Link | 4,040,298 | 735,781 | 84.59% | |
| ckim-vqsr | Link | 3,657,090 | 680,757 | 84.31% | |
| anovak-vg | Link | 4,296,761 | 1,131,758 | 79.15% | |
| ebanks-nist | Link | 3,151,019 | 0 | 100.00% | [#4] |
| rlopez-custom | Link | 3,097,788 | 57,975 | 98.16% | [#5] |
| egarrison-hhga | Link | 3,086,242 | 66,955 | 97.88% | [#6] |
| amark-mixed | Link | 3,083,646 | 121,074 | 96.22% | [#6] |
| ciseli-custom | Link | 3,064,375 | 259,836 | 92.18% | [#6] |
[#4] This entry was not considered because the entry submitted the NIST benchmark set as the answer
[#5] This entry was not considered because the VCF files were not submitted within the challenge timeframe
[#6] These entries were not considered because they did not call variants across the whole genome
The fact that concordance is no higher than 91.44% overall is probably indicative of limitations in the sequencing portion of the experiment. Several factors can contribute to decreased concordance, including potentially different batch of starting material from Coriell, and, more importantly, different handling for library preparation. Even if the same library is used, there is also inherent variability in the way sequencing instruments process a library and produce base calls — including differences in coverage across the whole genome. Nevertheless, it is desirable for NGS pipelines to be somewhat robust to noise or other artifacts, to the extent possible. We would therefore like to recognize the entries that produced results with more than 90% concordance, assigning the highest-concordance and high-concordance badges.
The accuracy comparisons 1a, 1b and 2 are quantifying the similarity between participants' results and the NIST (Genome in a Bottle) v2.19 gold standard, within the confident regions provided by NIST/GiaB. Each comparison outputs several metrics, including recall, precision and f-measure.
Recall, or sensitivity, reflects the percentage of variants in the NIST/GiaB benchmark set that were exactly called by the challenge participant pipeline in a submitted dataset. Precision, or positive predictive value, reflects the percentage of called variants which match exactly the NIST/GiaB benchmark set. F-measure is the harmonic mean of recall and precision, and is sometimes used as a single combined metric for evaluating overall accuracy.
The tables below summarize the results across accuracy comparisons. For each metric, the highest value between accuracy comparisons 1a and 1b is shown (column R1, P1, F1), along with the value for accuracy comparison 2 (columns R2, P2, F2). In each table, the entries are ranked based on the sum of their deltas from the top entry, i.e. for recall based on (99.54% - R1) + (98.92% - R2).
| Recall | |||
|---|---|---|---|
| Label | R1 | R2 | Δ |
| dgrover-gatk | 99.54% | 98.92% | 0.00% |
| amark-mixed | 99.46% | 98.62% | 0.38% |
| raldana-sentieon | 99.51% | 98.54% | 0.41% |
| mmohiyuddin-mixed | 99.40% | 98.62% | 0.44% |
| jedwards-sentieon | 99.44% | 98.46% | 0.56% |
| ckim-dragen | 99.34% | 98.50% | 0.62% |
| astatham-gatk | 99.05% | 98.19% | 1.22% |
| asubramanian-gatk | 98.91% | 98.19% | 1.36% |
| egarrison-hhga | 98.91% | 98.08% | 1.47% |
| cchapple-mixed | 98.95% | 97.96% | 1.55% |
| jharris-gatk | 98.82% | 97.45% | 2.19% |
| xli-custom | 98.59% | 97.56% | 2.31% |
| ckim-genalice | 97.96% | 96.75% | 3.75% |
| ckim-gatk | 99.25% | 94.25% | 4.96% |
| mlin-fermikit | 97.32% | 93.81% | 7.33% |
| ciseli-custom | 95.68% | 94.56% | 8.22% |
| ckim-isaac | 95.26% | 93.40% | 9.80% |
| ckim-vqsr | 95.48% | 90.60% | 12.38% |
| anovak-vg | 93.55% | 89.99% | 14.92% |
| rlopez-custom | 99.43% | 98.51% | [#7] |
| ebanks-nist | 100.00% | 100.00% | [#8] |
| Precision | |||
|---|---|---|---|
| Label | P1 | P2 | Δ |
| dgrover-gatk | 99.71% | 99.62% | 0.00% |
| jharris-gatk | 99.54% | 99.61% | 0.18% |
| ckim-vqsr | 99.48% | 99.56% | 0.29% |
| raldana-sentieon | 99.43% | 99.60% | 0.30% |
| egarrison-hhga | 99.54% | 99.46% | 0.33% |
| ckim-isaac | 99.41% | 99.47% | 0.45% |
| jedwards-sentieon | 99.35% | 99.47% | 0.51% |
| astatham-gatk | 99.31% | 99.47% | 0.55% |
| mmohiyuddin-mixed | 99.22% | 99.43% | 0.68% |
| cchapple-mixed | 99.25% | 99.39% | 0.69% |
| asubramanian-gatk | 99.12% | 99.39% | 0.82% |
| ckim-gatk | 99.21% | 99.29% | 0.83% |
| amark-mixed | 99.13% | 99.33% | 0.87% |
| ckim-dragen | 99.16% | 99.20% | 0.97% |
| mlin-fermikit | 98.63% | 97.90% | 2.80% |
| ckim-genalice | 98.11% | 97.76% | 3.46% |
| xli-custom | 97.86% | 97.97% | 3.50% |
| ciseli-custom | 93.16% | 95.26% | 10.91% |
| anovak-vg | 90.17% | 89.00% | 20.16% |
| rlopez-custom | 99.78% | 99.70% | [#7] |
| ebanks-nist | 100.00% | 100.00% | [#8] |
| F-measure | |||
|---|---|---|---|
| Label | F1 | F2 | Δ |
| dgrover-gatk | 99.62% | 99.27% | 0.00% |
| raldana-sentieon | 99.47% | 99.06% | 0.36% |
| jedwards-sentieon | 99.39% | 98.97% | 0.53% |
| mmohiyuddin-mixed | 99.31% | 99.02% | 0.56% |
| amark-mixed | 99.29% | 98.97% | 0.63% |
| ckim-dragen | 99.25% | 98.85% | 0.79% |
| astatham-gatk | 99.18% | 98.82% | 0.89% |
| egarrison-hhga | 99.22% | 98.76% | 0.91% |
| asubramanian-gatk | 99.00% | 98.79% | 1.10% |
| cchapple-mixed | 99.10% | 98.67% | 1.12% |
| jharris-gatk | 99.18% | 98.52% | 1.19% |
| xli-custom | 98.23% | 97.76% | 2.90% |
| ckim-gatk | 99.23% | 96.70% | 2.96% |
| ckim-genalice | 98.04% | 97.25% | 3.60% |
| mlin-fermikit | 97.97% | 95.81% | 5.11% |
| ckim-isaac | 97.29% | 96.34% | 5.26% |
| ckim-vqsr | 97.44% | 94.87% | 6.58% |
| ciseli-custom | 94.40% | 94.91% | 9.58% |
| anovak-vg | 91.83% | 89.49% | 17.57% |
| rlopez-custom | 99.61% | 99.10% | [#7] |
| ebanks-nist | 100.00% | 100.00% | [#8] |
[#7] This entry was not considered because the VCF files were not submitted within the challenge timeframe
[#8] This entry was not considered because the entry submitted the NIST benchmark set as the answer
Looking at recall, we noticed that most entries did better at Recall1 (Garvan) than Recall2 (HLI) — sometimes with striking differences (such as the entry labeled "macrogen-gatk"). This may be related to the fact that the Garvan dataset used newer, improved chemistry. We would like to recognize the top 6 entries (sum of deltas from the top less than 1%) assigning the highest-recall and high-recall badges.
A higher precision value means fewer false positives. Filtering criteria and other post-processing techniques are often used to reduce false positives, but sometimes this is at the expense of recall. Pipelines often have to deal with such tradeoffs between recall (sensitivity) and precision (positive predictive value), and different NGS pipelines may be tuned for different goals, depending on the context. Looking at the respective table for precision, the overall good performance of most entries (compared to the recall table) reveals that the majority of pipelines favor precision, especially since it does not show as big of a fluctuation between Precision1 (Garvan) and Precision2 (HLI). We would like to recognize the top 14 entries (sum of deltas from the top less than 1%) assigning the highest-precision and high-precision badges.
Given the performances in precision and recall in the first two tables, it's no surprise that the f-measure is usually slightly higher in the Garvan dataset, and that overall the entries are close (but not as close as in the precision table). We would like to recognize the top 11 entries (sum of deltas from the top less than 1.2%) assigning the highest-f-measure and high-f-measure badges.
We would like to extend special recognitions to the following entries:
- The extra-credit recognition to the entry submitted by Deepak Grover (Sanofi-Genzyme), for answering the extra credit question.
- The pfda-apps recognition to the entry submitted by Mike Lin (DNAnexus Science), for contributing apps to precisionFDA and using them to generate the results of the entry.
- The heroic-effort recognition to the entry submitted by Adam Novak et. al (VGTeam), for using this challenge as an opportunity to push the boundaries of their pipeline. As they mentioned in their submission, "vg is a work in progress, and this challenge answer represents the first time it has ever been run on whole genomes. The team sprinted through the weekend preceding the deadline in order to scale up the graph read-mapping and variant-calling algorithms, which had previously been used only on limited test regions".
- The promising recognition to the entry submitted by Rene Lopez et. al (MyBioinformatician). Although the submission was not received on time, the pipeline's performance characteristics were promising, and we are looking forward to seeing this method applied to future challenges.
This challenge used the comparison framework that was available when precisionFDA was first launched. The framework outputs detailed files with the variants of each category, so that anyone can perform downstream calculations or otherwise investigate their false positives or false negatives. This has helped participants better understand the characteristics of their pipelines – an effort that could be improved by more detailed reporting in the comparison output, since the current version of the comparison framework reports only aggregate statistics. As someone pointed out in a separate communication, “showing summary numbers for both SNPs and indels together can hide issues with indel calling behind the fact that SNPs do really well (most pipelines discover most of the easy SNPs)”. In the future, we look forward to more granular reporting, including separate statistics for SNPs versus indels and other types of variations, as well as information on zygosity mismatches. We are working with the GA4GH benchmarking group to incorporate the next generation of comparisons, and you can currently see an early glimpse of that in the GA4GH Benchmarking app on precisionFDA.
The challenge used the NIST/GiaB characterization of NA12878 as the benchmark set, as reported on GRCh37. Although NIST relied on different sequencing technologies and software algorithms to generate this dataset, members of the community expressed concerns that the dataset may be "biased towards GATK". Other community feedback mentioned known facts such as that the dataset is not comprehensive enough with respect to indels, that the confident regions are not covering several genomic areas of clinical importance, and that this dataset does not include copy-number variants or structural variants, or incorporate phasing information. GiaB is aware of the limitations of the existing dataset and is already working to address them. In addition, precisionFDA is collaborating with NIST to engage the community to improve this dataset by incorporating a Feedback button straight into the comparison results page. The government agencies, including FDA, NIST, CDC, and other stakeholders are also collaborating towards the generation of new benchmarking materials.
We also received questions around the choice of GRCh37. The performance of pipelines is affected by the reference human sequence, and as one participant put it “once we obtain a perfect (no gaps, no mistakes) reference genome, the performance will be much better”. The current NIST/GiaB release is based on GRCh37, but we are looking forward to using GRCh38 in the future.
This first challenge tried to engage the community, while at the same time making use of familiar datasets (such as NIST reference material) and simple evaluations (such as the comparison framework). The main focus was looking at reproducibility, while additionally assessing accuracy, both important concepts in regulatory science.
On one hand, as people said “if we all approach the challenge in good faith […], this challenge sets the right methodology: (1) examine the software repeatability between runs; (2) examine the reproducibility between datasets of the same DNA sample […]; (3) examine the accuracy against a well-characterized truth set.”; on the other hand, as someone else pointed out “a winning entry for this challenge would not necessarily (and in fact is unlikely) to work well on a different sample.”
This challenge focused on the consistency of the results obtained from both a single as well as two different runs of the same sample. Although the sample that was used has known “truth data” attached to it for certain kinds of calls (“high confidence variant calls”), so that the community can at least partially assess the accuracy of the results obtained, the precisionFDA team was primarily focused on the reproducibility. Part of the reasoning behind that has been very eloquently stated by some of the challenge participants – with so few samples that have known “truth” it is very easy to over-fit the results, so “accuracy” obtained does not provide the complete picture. But we also heard, “This is the first time that software repeatability and inter-dataset reproducibility have been put at front and center of judging a genomics pipeline’s quality. Without repeatability and reproducibility, accuracy is at best a one-time stroke of luck”. We are, however, taking a second look at accuracy in the precisionFDA Truth Challenge.
Lastly, we acknowledge that the evaluation of the results was confined within certain limits; however, the process (and its limitations) can inform future challenges. For example, we did not factor pipeline runtime or resource consumption into our evaluations.
We want to thank those of you who participated in our first challenge! By participating and putting your results and your thoughts out in the public, you fulfilled the first and most important goal of this challenge – to engage and get the dialogue started.
This challenge created a dataset that people can study further, if they so choose. We have created an archive of 60 VCF files (compressed with bgzip, and accompanied by tabix index files) that is available as a file on precisionFDA. We have also created a similar app asset, which can be included in apps that need direct access to the unarchived VCF files. We are excited to see how the community will use this in the future. Perhaps the results available from the challenge can provide some interesting ideas for precisionFDA and the scientific community to build upon.
In closing, we would like to leave you with several encouraging quotes from our community, which will hopefully inspire all readers to participate in one of our upcoming challenges:
“When I started working in this industry I felt that everyone claimed that their pipeline was better, but there was no objective way of measuring, so it sounded like an ad for car insurance. I think your challenges will move our industry forward immensely.”
“I wish to congratulate your team for putting together a bioinformatics challenge like that, it is a nice and unique idea. It can become a significant community resource for NGS work and data in future”.
“The challenge made me aware of many good software methods, thanks!”
“We found this experience is very helpful for us to improve our understanding of our pipeline.”
“We plan on contacting a fellow participant to try their pipeline.”