PrecisionFDA
Consistency Challenge
Engage and improve DNA test results with our first community challenge
Challenge Time Period
February 25, 2016 through April 25, 2016
At a glance
In the context of whole human genome sequencing, software pipelines typically rely on mapping sequencing reads to a reference genome and subsequently identifying variants (differences). One way of assessing the performance of such pipelines is by using well-characterized datasets such as Genome in a Bottle’s NA12878.
By supplying NA12878 whole-genome sequencing read datasets (FASTQ), and a framework for comparing variant call format (VCF) results, this challenge provides a common frame of reference for measuring some of the aspects of reproducibility and accuracy of participants’ pipelines.
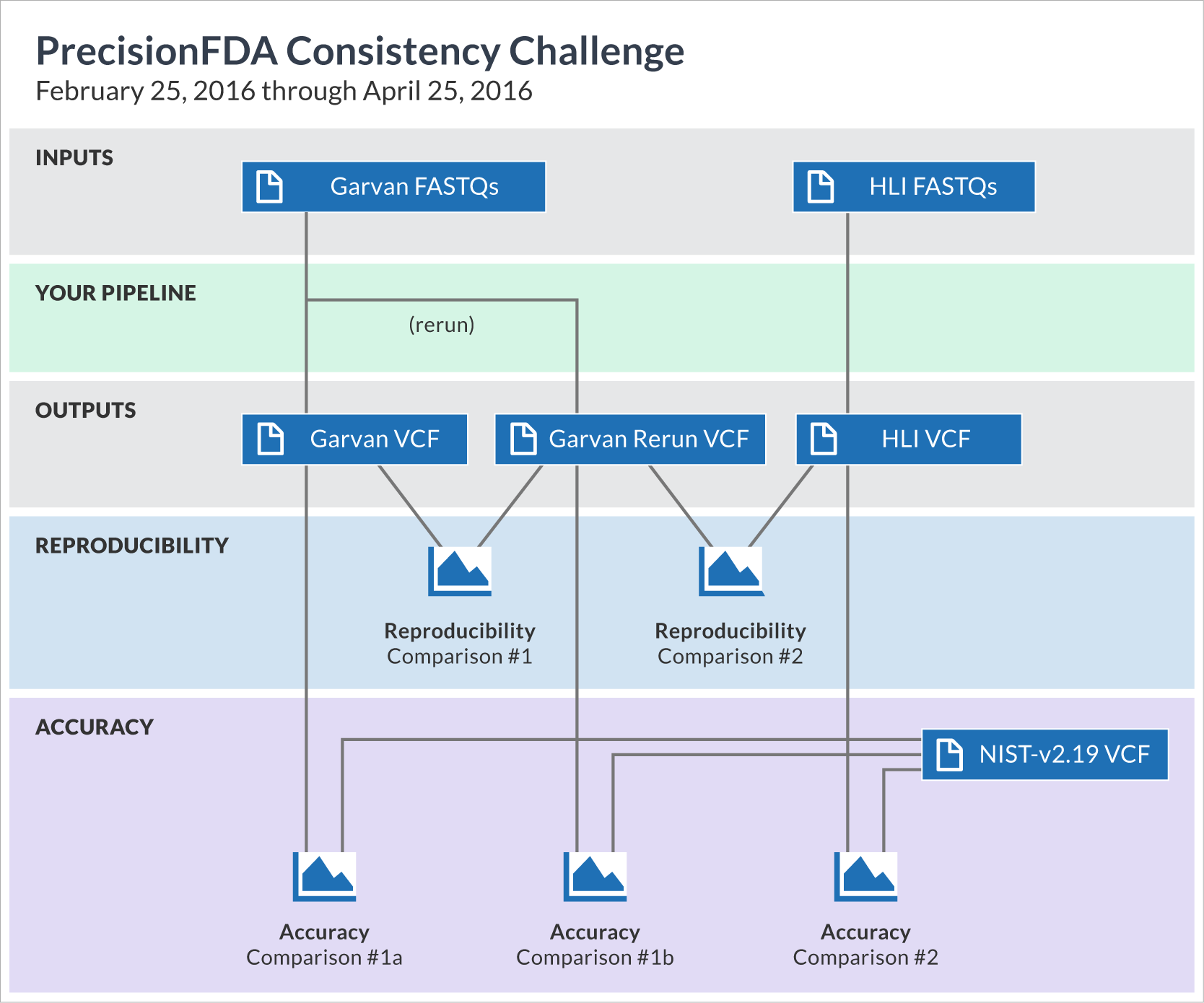
The challenge begins with two precisionFDA-provided input datasets, corresponding to whole-genome sequencing of the NA12878 human sample at two different sequencing sites. Your mission is to process these FASTQ files through your mapping and variation calling pipeline and create VCF files. For one of the datasets, you are required to do a rerun of your pipeline and obtain a rerun VCF as well. You can generate those results on your own environment, and upload them to precisionFDA, or you can reconstruct your pipeline on precisionFDA and run it on the cloud.
Regardless of how you generate your VCF files, you will subsequently use the precisionFDA comparison framework to conduct several pairwise comparisons:
- By comparing the rerun VCF to the original one, you will evaluate your pipeline’s reproducibility with respect to the same exact input file.
- By comparing the VCF files of the two datasets, you will evaluate reproducibility on the same sample across different sites.
- By comparing each of your three VCF files to the NIST (Genome in a Bottle) benchmark VCF, you will get estimates for accuracy.
The complete set of these five comparisons constitutes your submission entry to the challenge. Each comparison outputs several metrics (such as precision*, recall*, f-measure, or number of non-common variants). Selected participants and winners** will be recognized on the precisionFDA website. Therefore, we hope you are willing to share your experience with others to further enhance the community's effort to ensure consistency of tests.
The challenge runs until April 25, 2016.
Challenge details
Last updated: March 2nd, 2016
If you do not yet have a contributor account on precisionFDA, file an access request with your complete information, and indicate that you are entering the challenge. The FDA acts as steward to providing the precisionFDA service to the community and ensuring proper use of the resources, so your request will be initially pending. In the meantime, you will receive an email with a link to access the precisionFDA website in browse (guest) mode. Once approved, you will receive another email with your contributor account information.
With your contributor account you can use the features required to participate in the challenge (such as transfer files or run comparisons). Everything you do on precisionFDA is initially private to you (not accessible to the FDA or the rest of the community) until you choose to publicize it. So you can immediately start working on the challenge in private, and whenever you are ready you can officially publish your results as your challenge entry.
The starting point for this challenge consists of two datasets, corresponding to whole-genome sequencing of the NA12878 sample on an Illumina HiSeq X Ten instrument at two different sites (Garvan and HLI). A pair of gzipped FASTQ files is provided for each dataset. The following table summarizes key information for each dataset:
| Dataset/Site | Garvan | HLI |
|---|---|---|
| Contributor | Garvan Institute of Medical Research | Human Longevity, Inc. |
| Files |
NA12878-Garvan-Vial1_R1.fastq.gz
NA12878-Garvan-Vial1_R2.fastq.gz |
TSNano_1lane_L008_13801_NA12878_R1_001.fastq.gz
TSNano_1lane_L008_13801_NA12878_R2_001.fastq.gz |
| Starting Material | Coriell NA12878 DNA, lot NA12878*I2 | |
| Library Prep | TruSeq Nano DNA Library Prep kit, v2.5, with no sample multiplexing | TruSeq Nano NA12878 library, sequenced with the original V1 chemistry |
| Read Length | 2x150bp | 2x150bp |
| Insert Size | 350bp | 319bp |
| Instrument | HiSeq X (one lane) | HiSeq X (one lane) |
We would like to thank the Garvan Institute of Medical Research, and Human Longevity, Inc., for contributing these input files.
In the next step you will need to process these FASTQ files through your pipeline to generate VCF files. This can be done either by downloading the files and running your pipeline on your own environment, or by reconstructing your pipeline on precisionFDA and running it on the cloud. If you will be working on your own environment, download these datasets by visiting the links above and clicking the Download button (web-browser download, not recommended for large files) or the Authorized URL button.
After familiarizing yourself with the input files, you will need to process them through your mapping and variation calling pipeline to generate corresponding VCF files. Your pipeline must call variants across the whole genome. Each invocation of your pipeline must take as input a pair of FASTQ files and produce a VCF file containing exactly one genotyped sample. Results must be reported on GRCh37 human coordinates (i.e. chromosomes named 1, 2, ..., X, Y, and MT). You are strongly encouraged to compress each VCF file with bgzip, to reduce the file size.
| DO | DON'T |
|---|---|
|
Use GRCh37 Call variants across the whole genome Compress with bgzip |
Use hg19 or GRCh38 Call variants only in specific regions Generate a gVCF |
You must invoke your pipeline three times: 1) invoke it on the Garvan pair of FASTQ files to obtain a Garvan VCF; 2) re-invoke it on the same Garvan pair of FASTQ files to obtain a Garvan Rerun VCF; 3) invoke it on the HLI pair of FASTQ files to obtain an HLI VCF.
| Invocation | Input dataset (FASTQ pair) | Output dataset | Example output filename |
|---|---|---|---|
| #1 | Garvan | Garvan VCF | YourName-Garvan.vcf.gz |
| #2 | Garvan | Garvan Rerun VCF | YourName-Garvan-rerun.vcf.gz |
| #3 | HLI | HLI VCF | YourName-HLI.vcf.gz |
As discussed above, you are requested to repeatedly run your pipeline on the same input (for the Garvan pair of FASTQ files). Perfectly deterministic methods are expected to produce the same results when run on the same inputs; under such circumstances, it’s possible for the Garvan VCF to be identical to the Garvan Rerun VCF.
If you are running your pipeline in your own environment, upload the three generated files to precisionFDA. Additional information on uploading files is available at the precisionFDA docs. Your uploaded files are private, until you are ready to share them with the community (see "Submitting your entry" below).
Besides running your pipeline in your own environment, you have the additional option of reconstructing your pipeline on precisionFDA and running it on the cloud. To do that, you must create one or more apps on precisionFDA that encapsulate the actions performed in your pipeline. To create an app, you can provide Linux executables and an accompanying shell script to be run inside an Ubuntu VM on the cloud. The precisionFDA website contains extensive documentation on how to create apps, and you can also click the Fork button on an existing app (such as bwa_mem_bamsormadup) to use it as starting point for developing your own.
Constructing your pipeline on precisionFDA has an important advantage: you can, at your discretion, share it with the community, so that others can take a look at it and reproduce your results – and perhaps build upon it and further improve it.
Regardless of how you generate your VCFs (whether in your own environment or directly on precisionFDA), the next step is to conduct a total of five comparisons to evaluate aspects of reproducibility and accuracy.
The precisionFDA comparison framework makes use of vcfeval by Real Time Genomics (Cleary et al., 2015) to perform a pairwise comparison between a test VCF and a benchmark VCF, optionally constrained within certain coordinates provided by BED files (a test BED and a benchmark BED). For more information consult the precisionFDA docs. The following table summarizes the required comparisons:
| Comparison | Test VCF | Test BED | Benchmark VCF | Benchmark BED |
|---|---|---|---|---|
| Reproducibility Comparison #1 | Garvan VCF | - | Garvan Rerun VCF | - |
| Reproducibility Comparison #2 | Garvan VCF (or Garvan Rerun VCF) | - | HLI VCF | - |
| Accuracy Comparison #1a | Garvan VCF | - | NIST v2.19 VCF | NIST v2.19 BED |
| Accuracy Comparison #1b | Garvan Rerun VCF | - | NIST v2.19 VCF | NIST v2.19 BED |
| Accuracy Comparison #2 | HLI VCF | - | NIST v2.19 VCF | NIST v2.19 BED |
Reproducibility comparison #1
- is between your Garvan VCF and your Garvan Rerun VCF;
- it is meant to evaluate the reproducibility of your pipeline when running it repeatedly on the same FASTQ input.
Reproducibility comparison #2
- is between your Garvan VCF (or, at your discretion, its Rerun) and your HLI VCF;
- it is meant to evaluate reproducibility across different sequencing sites, where the same sequencing instrument and sample were used (but with potentially different sequencing conditions and parameters).
Accuracy comparisons
- are between each of your VCF files and the NIST v2.19 (Genome in a Bottle) benchmark VCF file, constrained within the coordinates of the accompanying NIST v2.19 BED file.
- The precisionFDA website provides NA12878-NISTv2.19.vcf.gz (to be used as benchmark VCF) and NA12878-NISTv2.19.bed (to be used as benchmark BED).
- These comparisons are meant to estimate the accuracy of your pipeline within the “confident” regions of the Genome in a Bottle “gold standard” dataset – therefore you must leave the test BED entry blank.
Your comparisons are private, until you are ready to share them with the community (see "Submitting your entry" below).
This challenge has been posted to the precisionFDA “Discussions” section. Discussion answers can include rich text formatting, as well as attachments such as files or comparisons. You can leverage this functionality to submit your entry by filling in your answer to the question.
In your answer’s text, please identify whether you are participating as an individual or as part of the team (and, if it is a team effort, don’t forget to identify the members of your team). Write a description of the pipeline used to obtain the results, and identify the name, version and command-line parameters of the mapper and variant caller invoked in your pipeline.
If you opted to construct your pipeline on precisionFDA, and your VCFs were generated by running one or more precisionFDA apps, the system will automatically prompt you to share the details of these executions when publishing your answer (see below). In that case, your text only needs to provide a high-level summary, because the exact software invocations will be available to the community via the system’s sharing mechanism.
Once you save your answer, it is initially private (and only visible to you). You can go back at any point and edit it, so we encourage you to start drafting it right away, and revise it as you make progress. Once you instantiate your answer, you can attach your comparisons to it – visit each of your comparisons and click Attach, then select your answer. Your entry must contain the five comparisons (as outlined in the previous section) in order to be considered valid.
Once you are comfortable with your answer, you can publish it so that others can see it (and don’t worry – you can still edit it or update its attachments even after publication). When publishing, the system will ask if you want to publish the comparisons, as well as the VCF files that were input to the comparisons; please choose that option to ensure that these artifacts are shared with the precisionFDA community. Entries that do not have their comparisons and VCF files published will not be considered valid. If you used apps on precisionFDA (instead of generating these VCF files externally), please choose the option to share the underlying jobs, apps, and app assets.
PrecisionFDA will select a number of entries which will receive an acknowledgement for participation in the challenge. Among the selected entries, winners** will be recognized on the precisionFDA website, for achievements across categories such as:
Reproducibility comparison #1
- Lowest count of unique/non-reproducible variants (“false positives” + “false negatives”).
Reproducibility comparison #2
- Lowest count of unique/non-reproducible variants (“false positives” + “false negatives”)
Accuracy comparisons #1a/#1b
Accuracy comparison #2
For more information about these metrics (precision, recall, f-measure, etc.) consult the precisionFDA docs.
It is possible for a pipeline to try to reach maximum precision* by being trivially strict, or maximum recall* by being trivially lenient. To avoid very extreme entries, and to ensure fairness in the competition, the challenge requires that all submitted accuracy comparisons reach a minimum threshold of 90% for each of the precision* and the recall* statistics. Entries that do not meet these criteria or are otherwise not deemed valid will not be considered.
Share your thoughts with the community (and any relevant software) on the following topic, and be acknowledged on the precisionFDA website as a precisionFDA Innovation Leader.
Comparing VCF results is a difficult problem (partly due to the fact that the same variant can be represented in many ways), but becomes more tractable for the case of a pairwise comparison. The reproducibility portion of this challenge leverages the precisionFDA pairwise comparison framework to assess the similarity between two pipeline invocations at a time, but extending it beyond two trials may not be as straightforward.
How would you assess reproducibility across more than two repeated experiments? What is an ideal method for comparing multiple (more than two) VCF files? How tractable is a seemingly simple calculation such as “count the number of variants that are common (reproducible) among a set of VCF files”? Please include any software solutions, links to tools or scripts, etc.
Footnotes
* The terminology currently used in the precisionFDA comparison output (such as "precision" and "recall") is not necessarily harmonized with definitions used by ISO, CLSI, or FDA, but are terms commonly used by NGS software developers.
** Winning a precisionFDA challenge is an acknowledgement by the precisionFDA community and does not imply FDA endorsement of any organization, tool, software, etc.