PrecisionFDA
Truth Challenge
Engage and improve DNA test results with our community challenges
- Ryan Poplin
- Mark DePristo
- Verily Life Sciences Team
- Rafael Aldana
- Hanying Feng
- Brendan Gallagher
- Jun Ye
for Clinical Genomics
- Aaron Statham
- Mark Cowley
- Joseph Copty
- Mark Pinese
- Deepak Grover
- Deepak Grover
- Rafael Aldana
- Hanying Feng
- Brendan Gallagher
- Jun Ye
We are very excited to see growing numbers of challenge participants! For this challenge we received 35 entries, not only from many who participated in the previous challenge, but also from several first time participants. Some participants showcased their developing new methods, while others decided to use existing methods and see how they perform in this particular environment. We are extremely grateful to all participants for their willingness to share their results and to contribute to this effort.
This second precisionFDA challenge was conducted in collaboration with the Genome in a Bottle (GiaB) consortium, which provided the "truth" data, as well as with the Global Alliance for Genomics and Health (GA4GH), which provided best practices and software for conducting the comparison of participants' entries to the truth data. In fact, this effort represents the first time that this new truth data has been investigated (and it's only an approximation of the truth, hence sometimes we say "truth" instead of truth), and the first time that this new comparison methodology has been applied at scale across the vast number of submitted entries.
Our evaluation against the HG002 truth data has produced several metrics (such as f-score, recall and precision) across different variant types (such as SNP and indels), subtypes (such as insertions or deletions of specific size ranges) and genomic contexts (such as whole genome, coding regions, etc.), leading to thousands of computed numbers per challenge entry. We've chosen six of them (f-score, recall and precision, across SNPs and across indels, in the whole genome) to use as the basis for handing out awards and recognitions.
These results are by no means the final word. Given the originality of the truth data and the comparison methodology, we expect the community to further conduct analyses and contribute to improvements in the benchmarking methodology, the correctness of the truth data, and the definition of comparison metrics.
We would like to acknowledge and thank all of those who participated in the precisionFDA Truth Challenge. As with the previous challenge, we hope that everyone will feel like a winner.
The following table summarizes the challenge entries, and the results of the comparison against the HG002 truth data.
We've given each entry a unique label, comprised of the name of the submitting user as well as a short mnemonic keyword representing the pipeline. (As with the previous challenge, these keywords are merely indicative of each pipeline's main component, hence somewhat subjective; for a more faithful description of each pipeline, refer to the full text that accompanied each submission by following the label links). Each submitted entry consisted of two VCFs, corresponding to the variants called on the HG001/NA12878 and HG002/NA24385 datasets respectively. Links to these files can be found in the "Datasets" tab of the table.
The entries are sorted alphabetically. You can click on any column header to re-sort the table according to that column.
| Label | Submitter | Organization | SNP-Fscore | SNP-recall | SNP-precision | INDEL-Fscore | INDEL-recall | INDEL-precision |
|---|---|---|---|---|---|---|---|---|
| raldana-dualsentieon | Rafael Aldana et al. | Sentieon | 99.9260 | 99.9131 | 99.9389 | 99.1095 | 98.7566 | 99.4648 |
| bgallagher-sentieon | Brendan Gallagher et al. | Sentieon | 99.9296 | 99.9673 | 99.8919 | 99.2678 | 99.2143 | 99.3213 |
| mlin-fermikit | Mike Lin | DNAnexus Science | 98.8629 | 98.2311 | 99.5029 | 95.5997 | 94.8918 | 96.3183 |
| jmaeng-gatk | Ju Heon Maeng | Yonsei University | 99.6144 | 99.4608 | 99.7686 | 99.1098 | 99.0216 | 99.1981 |
| ckim-dragen | Changhoon Kim | Macrogen | 99.8268 | 99.9524 | 99.7015 | 99.1359 | 99.1574 | 99.1143 |
| ckim-gatk | Changhoon Kim | Macrogen | 99.6466 | 99.4788 | 99.8150 | 99.2271 | 99.1551 | 99.2992 |
| ckim-isaac | Changhoon Kim | Macrogen | 98.5357 | 97.1616 | 99.9494 | 95.8099 | 93.7006 | 98.0163 |
| ckim-vqsr | Changhoon Kim | Macrogen | 99.2866 | 98.6511 | 99.9303 | 99.2541 | 99.0614 | 99.4476 |
| ltrigg-rtg2 | Len Trigg | RTG | 99.8749 | 99.8935 | 99.8562 | 99.2539 | 98.8759 | 99.6347 |
| ltrigg-rtg1 | Len Trigg | RTG | 99.8754 | 99.8921 | 99.8587 | 99.0160 | 98.3355 | 99.7061 |
| hfeng-pmm1 | Hanying Feng et al. | Sentieon | 99.9496 | 99.9227 | 99.9766 | 99.3397 | 99.0289 | 99.6526 |
| hfeng-pmm2 | Hanying Feng et al. | Sentieon | 99.9416 | 99.9254 | 99.9579 | 99.3119 | 99.0152 | 99.6103 |
| hfeng-pmm3 | Hanying Feng et al. | Sentieon | 99.9548 | 99.9339 | 99.9756 | 99.3628 | 99.0161 | 99.7120 |
| jlack-gatk | Justin Lack | NIH | 99.7200 | 99.9393 | 99.5016 | 98.6899 | 98.8138 | 98.5664 |
| astatham-gatk | Aaron Statham et al. | KCCG | 99.5934 | 99.2091 | 99.9807 | 99.3424 | 99.2404 | 99.4446 |
| qzeng-custom | Qian Zeng | LabCorp | 99.4966 | 99.2413 | 99.7533 | 96.8316 | 96.8703 | 96.7929 |
| anovak-vg | Adam Novak et al. | vgteam | 98.4545 | 98.3357 | 98.5736 | 70.4960 | 69.7491 | 71.2591 |
| eyeh-varpipe | ErhChan Yeh et al. | Academia Sinica | 99.4670 | 99.9638 | 98.9751 | 92.5779 | 91.3854 | 93.8021 |
| ciseli-custom | Christian Iseli et al. | SIB | 97.7648 | 98.8356 | 96.7169 | 83.5453 | 82.5314 | 84.5844 |
| ccogle-snppet* | Christopher Cogle et al. | CancerPOP | ||||||
| asubramanian-gatk | Ayshwarya Subramanian et al. | Broad Institute | 98.9379 | 97.9985 | 99.8954 | 98.8418 | 98.5404 | 99.1451 |
| rpoplin-dv42 | Ryan Poplin et al. | Verily Life Sciences | 99.9587 | 99.9447 | 99.9728 | 98.9802 | 98.7882 | 99.1728 |
| cchapple-custom | Charles Chapple et al. | Saphetor | 99.8448 | 99.8832 | 99.8063 | 99.1388 | 98.8448 | 99.4346 |
| gduggal-bwafb | Geet Duggal et al. | DNAnexus Science | 99.7820 | 99.8619 | 99.7021 | 96.9474 | 95.5004 | 98.4390 |
| gduggal-bwaplat | Geet Duggal et al. | DNAnexus Science | 98.8646 | 98.0471 | 99.6958 | 92.6621 | 87.0843 | 99.0034 |
| gduggal-bwavard | Geet Duggal et al. | DNAnexus Science | 99.3249 | 99.0431 | 99.6083 | 87.3464 | 87.1769 | 87.5166 |
| gduggal-snapfb | Geet Duggal et al. | DNAnexus Science | 99.2501 | 99.8026 | 98.7037 | 92.2602 | 90.5733 | 94.0112 |
| gduggal-snapplat | Geet Duggal et al. | DNAnexus Science | 99.0030 | 98.6815 | 99.3266 | 76.4210 | 69.0418 | 85.5664 |
| gduggal-snapvard | Geet Duggal et al. | DNAnexus Science | 99.0871 | 98.9341 | 99.2406 | 83.0264 | 83.4429 | 82.6139 |
| ghariani-varprowl | Gunjan Hariani et al. | Quintiles | 99.3496 | 99.8685 | 98.8361 | 87.2025 | 87.3272 | 87.0781 |
| jpowers-varprowl | Jason Powers et al. | Q2 Solutions | 99.5004 | 99.5447 | 99.4561 | 86.4885 | 85.2886 | 87.7226 |
| dgrover-gatk | Deepak Grover | Sanofi-Genzyme | 99.9456 | 99.9631 | 99.9282 | 99.4009 | 99.3458 | 99.4561 |
| egarrison-hhga | Erik Garrison et al. | - | 99.8985 | 99.8365 | 99.9607 | 97.4253 | 97.1646 | 97.6874 |
| jli-custom | Jian Li et al. | Roche | 99.9382 | 99.9603 | 99.9160 | 99.3675 | 99.0788 | 99.6580 |
| ndellapenna-hhga | Nicolas Della Penna | ANU | 99.8818 | 99.8118 | 99.9519 | 97.3838 | 97.0938 | 97.6756 |
For more information about how these results were calculated, consult the comparison section below.
We are handing out the following community challenge awards:
- highest-snp-performance to the entry submitted by Ryan Poplin et al. from Verily Life Sciences, for achieving the highest SNP F-score.
- highest-snp-recall to the entry submitted by Brendan Gallagher et al. from Sentieon (labeled bgallagher-sentieon), for achieving the highest SNP recall.
- highest-snp-precision to the entry submitted by Aaron Statham et al. from Kinghorn Center for Clinical Genomics, for achieving the highest SNP precision.
- highest-indel-performance to the entry submitted by Deepak Grover from Sanofi-Genzyme, for achieving the highest indel F-score.
- highest-indel-recall to the entry submitted by Deepak Grover from Sanofi-Genzyme, for achieving the highest indel recall.
- highest-indel-precision to the entry submitted by Hanying Feng et al. from Sentieon (labeled hfeng-pmm3), for achieving the highest indel precision.
We are also handing out the following recognitions:
- high-snp-performance to all entries achieving a SNP F-score of 99.920% or higher.
- high-snp-recall to all entries achieving a SNP recall of 99.910% or higher.
- high-snp-precision to all entries achieving a SNP precision of 99.920% or higher.
- high-indel-performance to all entries achieving an indel F-score of 99.310% or higher.
- high-indel-recall to all entries achieving an indel recall of 99.150% or higher.
- high-indel-precision to all entries achieving an indel precision of 99.430% or higher.
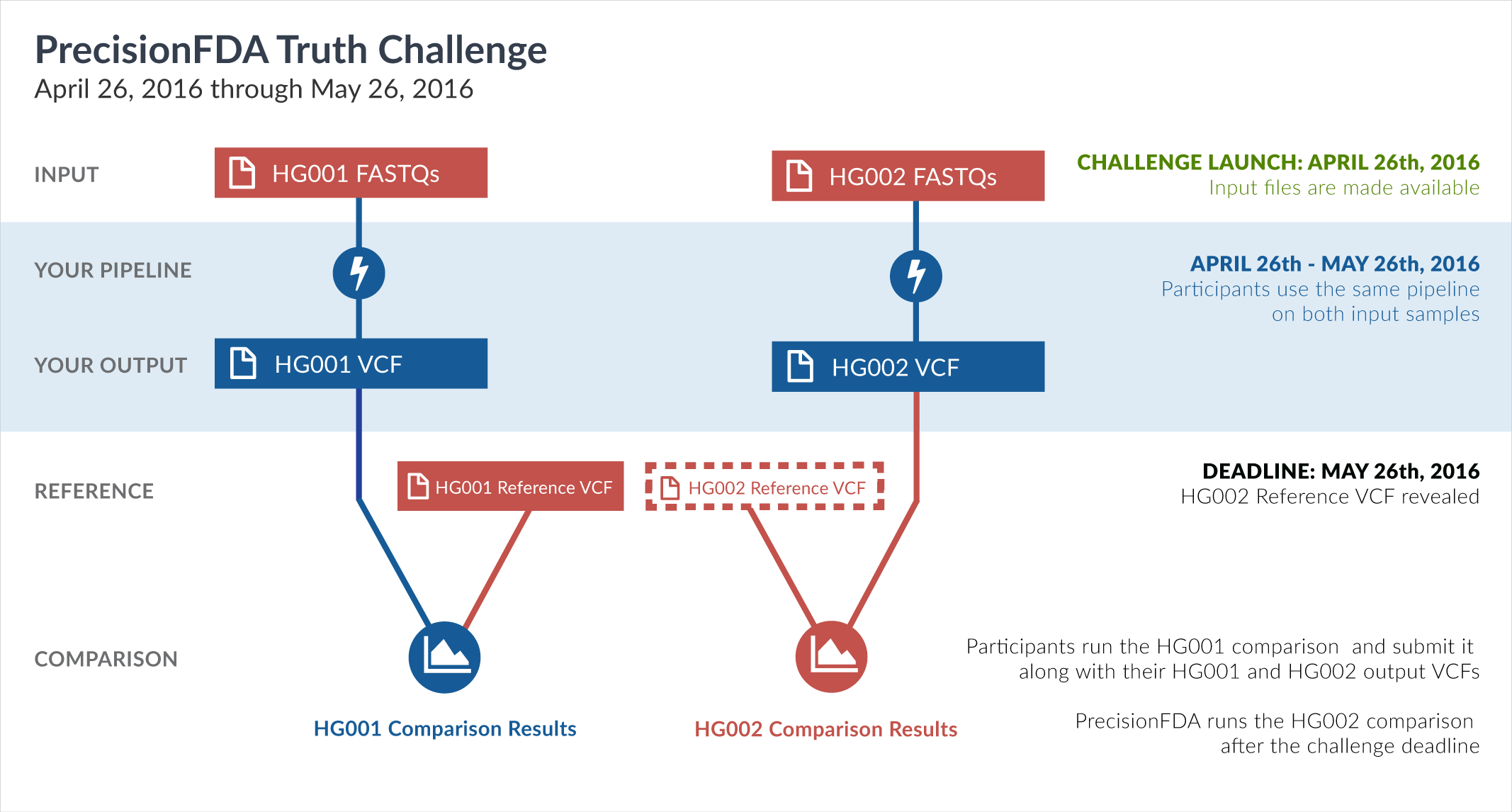
One way of learning about the performance of software pipelines for processing human genome sequencing data is by using well-characterized datasets such as NA12878/HG001. The previous challenge made use of the GiaB/NIST NA12878 v2.19 truth data as part of the evaluation of accuracy. But considering how widely HG001 has been used to train software pipelines, this challenge focuses on a new sample, HG002, whose truth data was not yet available at the time of the challenge.
In particular, this challenge provided sequencing reads datasets (FASTQ) for both HG001 and HG002. During the challenge, participants had access to the aforementioned HG001 v2.19 truth data, but not to the HG002 truth data. Upon the closing of the Truth Challenge, the Genome in a Bottle (GiaB) consortium, led by the National Institute of Standards (NIST), released the HG002 truth data, as well as an updated version of the HG001 truth data. Both were initially assigned version 3.2.
The truth data files have been published on precisionFDA by Dr. Justin Zook (scientist at the National Institute of Standards and Technology and co-leader of the Genome in a Bottle Consortium efforts), in the precisionFDA discussion topic titled Help improve the new v3.2 GIAB benchmark calls for HG001 and HG002.
For your convenience, we've included below a copy of the announcement. It discusses known issues of the truth data as well as other important caveats, and we encourage you to read it to understand how this new truth data may have affected the results of the challenge, or may interact with your own evaluations in the future.
NEW RESULTS
Based on feedback received about our v3.2 calls, we have now uploaded a new version 3.2.2 set of calls that fixes a significant number of erroneously low confidence variants on chr7 and chr9, and also now excludes regions homologous to the hs37d5 decoy sequence.
Changes in v3.2.2: We found a bug in one of the input call sets that was causing many of the variants on chr7 in HG001 and HG002 and on chr9 on HG002 to be reported as not high confidence. We have corrected this issue, and all other chromosomes are the same as in v3.2.1.
Changes in v3.2.1: The only change in v3.2.1 is to exclude regions from our high confidence bed file if they have homology to the decoy sequence hs37d5. These decoy-related regions were generated by Heng Li and are available as mm-2-merged.bed at https://github.com/ga4gh/benchmarking-tools/tree/master/resources/stratification-bed-files/SegmentalDuplications. Our high-confidence vcf generally does not include variants if they are homologous to decoy sequence. Although most of these likely should be excluded, some real variants may be missed, so we are excluding these regions from our high-confidence regions until they are better characterized. However, it is important to note that these regions may be enriched for false positives if the decoy is not used in mapping.
Description of v3.2: We have been developing a new, reproducible integration process to create high-confidence SNP, small indel, and homozygous reference calls for benchmarking small variant calls. We have used this new process to generate v3.2 of our high-confidence calls for HG001 (aka NA12878), as well as the first high-confidence callset for HG002 (aka Ashkenazim Jewish Son), which are attached to this discussion and under ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/release/. These calls were generated only from data generated from our NIST Reference Material batch of cells. Planned future work will include (1) generating similar callsets on the GIAB/PGP AJ parents and Chinese trio, (2) incorporating additional datasets from these genomes to characterize more difficult regions, (3) incorporating pedigree information for phasing and to improve the calls, (4) developing better methods for homopolymer characterization, (5) making calls on X and Y for males, and (6) characterizing larger indels and structural variation. We greatly appreciate the feedback many of you have given already, and we very much welcome additional feedback to help us improve future versions of these calls as comments in this discussion, and about specific challenging variants at http://goo.gl/forms/OCUnvDXMEt1NEX8m2.
We highly recommend reading the information below prior to using these calls to understand how best to use them and their limitations. There is also a draft google doc that describes the methods used to form these calls and some preliminary comparisons to other callsets at: https://docs.google.com/document/d/1NE-NGSMslFFndQpbqnvu4wfnwgmgOcQu6KIjSbCauYg/edit?usp=sharing
Best Practices for Using High-confidence Calls:
Benchmarking variant calls is a complex process, and best practices are still being developed by the Global Alliance for Genomics and Health (GA4GH) Benchmarking Team (https://github.com/ga4gh/benchmarking-tools/). Several things are important to consider when benchmarking variant call accuracy:
- Complex variants (e.g., nearby SNPs and indels or block substitutions) can often be represented correctly in multiple ways in the vcf format. Therefore, we recommend using sophisticated benchmarking tools like those developed by members of the GA4GH benchmarking team. Currently, the most developed tools are rtgtools vcfeval (http://realtimegenomics.com/products/rtg-tools/) or hap.py (https://github.com/Illumina/hap.py). See supplementary information in https://docs.google.com/document/d/1NE-NGSMslFFndQpbqnvu4wfnwgmgOcQu6KIjSbCauYg/edit?usp=sharing for a suggested process for doing comparisons.
- By their nature, high-confidence variant calls and regions tend to include a subset of variants and regions that are easier to characterize. Accuracy of variant calls outside the high-confidence regions is generally likely to be lower than inside the high-confidence regions, so benchmarking against high-confidence calls will usually overestimate accuracy for all variants in the genome. Similarly, it is possible that a variant calling method has higher accuracy statistics compared to other methods when compared to the high-confidence variant calls but has lower accuracy compared to other methods for all variants in the genome.
- Stratification of performance for different variant types and different genome contexts can be very useful when assessing performance, because performance often differs between variant types and genome contexts. In addition, stratification can elucidate variant types and genome contexts that fall primarily outside high-confidence regions. Standardized bed files for stratifying by genome context are available from GA4GH at https://github.com/ga4gh/benchmarking-tools/tree/master/resources/stratification-bed-files.
- Particularly for targeted sequencing, it is critical to calculate confidence intervals around statistics like sensitivity because there may be very few examples of variants of some types in the benchmark calls in the targeted regions. Manual curation of sequence data in a genome browser for a subset of false positives and false negatives is essential for an accurate understanding of statistics like sensitivity and precision. Curation can often help elucidate whether the benchmark callset is wrong, the test callset is wrong, both callsets are wrong, or the true answer is unclear from current technologies. If you find questionable or challenging sites, reporting them will help improve future callsets. You can report information about particular sites at http://goo.gl/forms/OCUnvDXMEt1NEX8m2.
Known issues with high-confidence calls:
There are a number of known issues in the current high-confidence callset, which we plan to address in future releases:
- Compound heterozygous sites are sometimes represented as a heterozygous deletion that overlaps with a heterozygous SNP. At these sites, the genotype “0/1” or “0|1” should not be interpreted as meaning that the “0” haplotype is reference. Hap.py and vcfeval (with the --ref-overlap flag) interpret these sites as intended.
- The callset currently only includes local phasing information from freebayes, which does not use the PS field, so that locally phased variants should not be assumed to be phased with variants that are phased at a distant location.
- The high-confidence calls have an increased FP, FN, and genotyping error rate for single base indels in homopolymers. Some of these sites have a low allele fraction variant in multiple technologies so that it is unclear whether the site is a systematic sequencing error or a true variant in a fraction of the cells. Some of these sites are listed in https://docs.google.com/spreadsheets/d/1kHgRLinYcnxX3-ulvijf2HrIdrQWz5R5PtxZS-_s6ZM/edit?usp=sharing
Differences between v2.19 and v3.2 integration methods:
The new v3.2 integration methods differ from the previous GIAB calls (v2.18 and v2.19) in several ways, both in the data used and the integration process and heuristics:
- Only 4 datasets were used, selected because they represent 4 sequencing technologies that were generated from the NIST RM 8398 batch of DNA. They are: 300x Illumina paired end WGS, 100x Complete Genomics WGS, 1000x Ion exome sequencing, and SOLID WGS.
- Mapping and variant calling algorithms designed specifically for each technology were used to generate sensitive variant callsets where possible: novoalign + GATK-haplotypecaller and freebayes for Illumina, the vcfBeta file from the standard Complete Genomics pipeline, tmap+TVC for Ion exome, and Lifescope+GATK-HC for SOLID. This is intended to minimize bias towards any particular bioinformatics toolchain.
- Instead of forcing GATK to call genotypes at candidate variants in the bam files from each technology, we generate sensitive variant call sets and a bed file that describes the regions that were callable from each dataset. For Illumina, we used GATK callable loci to find regions with at least 20 reads with MQ >= 20 and with coverage less than 2x the median. For Complete Genomics, we used the callable regions defined by the vcfBeta file and excluded +-50bp around any no-called or half-called variant. For Ion, we intersected the exome targeted regions with callableloci (requiring at least 20 reads with MQ >= 20). Due to the shorter reads and low coverage for SOLID, it was only used to confirm variants, so no regions were considered callable.
- A new file with putative structural variants was used to exclude potential errors around SVs. These were SVs derived from multiple PacBio callers and multiple integrated illumina callers using MetaSV. These make up a significantly smaller fraction of the calls and genome (~4.5%) than the previous bed, which was a union of all dbVar calls for NA12878 (~10%).
- Homopolymers >10bp in length, including those interrupted by one nucleotide different from the homopolymer, are now excluded from the high-confidence regions, because these were seen to have a high error rate for all technologies. This constitutes a high fraction of the putative indel calls, so more work is needed to resolve these.
- A bug that caused nearby variants to be missed in v2.19 is fixed in the new calls.
- The new vcf contains variants outside the high-confidence bed file. This enables more robust comparison of complex variants or nearby variants that are near the boundary of the bed file. It also allows the user to evaluate concordance outside the high-confidence regions, but these concordance metrics should be interpreted with great care.
- We now output local phasing information when it is provided in the Illumina freebayes calls.
Shortly after the initial publication of v3.2 of the truth data, there were two subsequent updates leading to v3.2.1 and v3.2.2. These updates introduced improvements to the truth data based on feedback from the community and from artifacts discovered in initial evaluations of the challenge entries. The final comparison results discussed on this web page are based on comparisons against the latest version of the truth data (v3.2.2).
It is important to note that the methodology for compiling the truth data has changed substantially since the release of v2.19 of the HG001 truth dataset, and therefore the same VCF file may rank differently in a comparison against HG001 v2.19 versus a comparison against HG001 v3.2.2. These changes are discussed in the section titled "Differences between v2.19 and v3.2 integration methods" in Dr. Zook's announcement.
We would like to thank the National Institute of Standards and Technology, and the Genome in a Bottle consortium, for the excellent collaboration in this precisionFDA challenge. As can be attested by them, creating a truth dataset is a complicated process. The published dataset, even with its latest v3.2.2 improvements, is only an approximation of the truth. We hope you will keep that in mind when interpreting the results of this challenge and that you will exercise caution, as the determination of truth is always an ongoing process.
The precisionFDA system includes an initial VCF comparison framework which was used in the first precisionFDA challenge. We asked participants to use that framework to compare their HG001 VCFs against the GiaB/NIST HG001 v2.19 truth data while the challenge was still ongoing, to ensure that their files were in good shape. However, for the final evaluation of the results of this challenge, we partnered with the Benchmarking Task team of the GA4GH Data Working Group, to employ an updated comparison framework.
The GA4GH benchmarking team has been discussing the difficulties of conducting proper VCF comparisons for several months now, and has devised metrics definitions, initial specifications, early software prototypes, and other benchmarking resources for implementing VCF comparison best practices. We approached the chair of the benchmarking group, Dr. Justin Zook, to consult with him on how to best use these resources in the context of this precisionFDA challenge.
The outcome of our collaboration is the utilization of a particular methodology for comparing VCFs and counting results, which was finalized and tailored for the needs of this challenge, and which we hope will form the basis of other comparisons in the future. It is available on precisionFDA as an app, titled Vcfeval + Hap.py Comparison. (Given the departure from the previous comparison technique, and until we have received enough feedback for this new method, the "Comparison" feature of precisionFDA has not yet been updated.)
This particular comparison framework consists of a specific version of Real Time Genomics' vcfeval, used for VCF comparison, and a specific version of Illumina's hap.py, used for quantification; together they form the prototype GA4GH benchmarking workflow, as illustrated here.
The vcfeval tool generates an intermediate VCF which is further quantified by hap.py. The "quantify" tool from hap.py counts and stratifies variants. It counts SNPs and INDELs separately and provides additional counts for subtypes of each variant (indels of various lengths, het / hom, stratification regions). In order to determine if a variant specifies SNPs, insertions or deletions, "quantify" performs an alignment of the corresponding REF and ALT alleles. This improves handling of MNPs/complex variants and provides results that are comparable between different methods.
The quantification process calculates the following metrics, in harmony with "Comparison Method 3" as defined in this upcoming GA4GH benchmarking spec update:
| Metric | Definition |
|---|---|
| TRUTH.TP | True positives, from the perspective of the truth data, i.e. the number of sites in the Truth Call Set for which there are paths through the Query Call Set that are consistent with all of the alleles at this site, and for which there is an accurate genotype call for the event. |
| QUERY.TP | True positives, from the perspective of the query data, i.e. the number of sites in the Query Call Set for which there are paths through the Truth Call Set that are consistent with all of the alleles at this site, and for which there is an accurate genotype call for the event. |
| TRUTH.FN | False negatives, i.e. the number of sites in the Truth Call Set for which there is no path through the Query Call Set that is consistent with all of the alleles at this site, or sites for which there is an inaccurate genotype call for the event. Sites with correct variant but incorrect genotype are counted here. |
| QUERY.FP | False positives, i.e. the number of sites in the Query Call Set for which there is no path through the Truth Call Set that is consistent with this site. Sites with correct variant but incorrect genotype are counted here. |
| FP.gt | The number of false positives where the non-REF alleles in the Truth and Query Call Sets match (i.e. cases where the truth is 1/1 and the query is 0/1 or similar). |
| Recall | TRUTH.TP / (TRUTH.TP + TRUTH.FN) |
| Precision | QUERY.TP / (QUERY.TP + QUERY.FP) |
| F-score | Harmonic mean of Recall and Precision |
Note that recall uses TRUTH.TP whereas precision uses QUERY.TP. For recall, TRUTH.TP counts the number of truth variants reproduced in the truth set representation, which is the same for all callers and should also be consistent with the confident regions (especially important around confident region boundaries and where variants can move between categories depending on how a variant caller decides to represent them). Using QUERY.TP for recall would have introduced problems where different methods create different representations (e.g. systematically calling MNPs as insertions + deletions can skew indel numbers in the query). For calculating precision, the tool uses QUERY.TP (i.e. the precision measures the relative number of wrong calls for all query calls).
The quantification process reports results with additional granularity, according to a few different dimensions, as outlined in the four tabs of the following table:
| Value | Meaning |
|---|---|
| SNP | SNP or MNP variants |
| INDEL | Indels and complex variants |
| Value | Meaning |
|---|---|
| * | Aggregate numbers of all subtypes |
ti | SNPs that constitute transitions |
tv | SNPs that constitute transversions |
I1_5 | Insertions of length 1-5 |
I6_15 | Insertions of length 6-15 |
I16_PLUS | Insertions of length 16 or more |
D1_5 | Deletions of length 1-5 |
D6_15 | Deletions of length 6-15 |
D16_PLUS | Deletions of length 16 or more |
C1_5 | Complex variants of length 1-5 |
C6_15 | Complex variants of length 6-15 |
C16_PLUS | Complex variants of length 16 or more |
| Value | Meaning |
|---|---|
| * | Aggregate numbers of all genotypes |
het | Only heterozygous variant calls (0/1 or similar genotypes) |
homalt | Only homozygous alternative variant calls (1/1 or similar genotypes) |
het | Only heterozygous alternative variant calls (1/2 or similar genotypes) |
| Value | Meaning |
|---|---|
| * | Aggregate numbers not limited to any subset (but still within confident regions) |
func_cds | Coding exons from RefSeq |
map_l100_m2_e1 | Regions in which 100bp reads map to >1 location with up to 2 mismatches and up to 1 indel |
map_l150_m0_e0 | Regions in which 150bp reads map to >1 location with up to 0 mismatches and up to 0 indels |
map_l150_m2_e1 | Regions in which 150bp reads map to >1 location with up to 2 mismatches and up to 1 indel |
map_l150_m2_e0 | Regions in which 150bp reads map to >1 location with up to 2 mismatches and up to 0 indels |
map_l100_m1_e0 | Regions in which 100bp reads map to >1 location with up to 1 mismatch and up to 0 indels |
map_l125_m1_e0 | Regions in which 125bp reads map to >1 location with up to 1 mismatch and up to 0 indels |
map_l250_m2_e1 | Regions in which 250bp reads map to >1 location with up to 2 mismatches and up to 1 indel |
map_l250_m0_e0 | Regions in which 250bp reads map to >1 location with up to 0 mismatches and up to 0 indels |
map_l150_m1_e0 | Regions in which 150bp reads map to >1 location with up to 1 mismatch and up to 0 indels |
map_l125_m2_e0 | Regions in which 125bp reads map to >1 location with up to 2 mismatches and up to 0 indels |
map_l100_m0_e0 | Regions in which 100bp reads map to >1 location with up to 0 mismatches and up to 0 indels |
map_l250_m1_e0 | Regions in which 250bp reads map to >1 location with up to 1 mismatch and up to 0 indels |
map_l125_m2_e1 | Regions in which 125bp reads map to >1 location with up to 2 mismatches and up to 1 indel |
map_l250_m2_e0 | Regions in which 250bp reads map to >1 location with up to 2 mismatches and up to 0 indels |
map_l125_m0_e0 | Regions in which 125bp reads map to >1 location with up to 0 mismatches and up to 0 indels |
map_l100_m2_e0 | Regions in which 100bp reads map to >1 location with up to 2 mismatches and up to 0 indels |
map_siren | Regions considered difficult to map by amplab SiRen |
tech_badpromoters | 1000 promoter regions with lowest relative coverage in Illumina, relatively GC-rich |
lowcmp_SimpleRepeat_homopolymer_gt10 | Homopolymers >10bp in length |
lowcmp_SimpleRepeat_triTR_11to50 | Exact 3bp tandem repeats 11-50bp in length |
lowcmp_Human_Full_Genome_TRDB_hg19_150331_TRgt6_lt51bp_gt95identity_merged | Tandem repeats with >6bp unit size and <51bp in length and >95% identity |
lowcmp_Human_Full_Genome_TRDB_hg19_150331_all_merged | All tandem repeats from TRDB with adjacent repeats merged |
lowcmp_SimpleRepeat_quadTR_11to50 | Exact 4bp tandem repeats 11-50bp in length |
lowcmp_Human_Full_Genome_TRDB_hg19_150331_TRlt7_gt200bp_gt95identity_merged | Tandem repeats with 1-6bp unit size and >200bp in length and >95% identity |
lowcmp_Human_Full_Genome_TRDB_hg19_150331_TRlt7_lt101bp_gt95identity_merged | Tandem repeats with 1-6bp unit size and <101bp in length and >95% identity |
lowcmp_SimpleRepeat_quadTR_gt200 | Exact 4bp tandem repeats >200bp in length |
lowcmp_Human_Full_Genome_TRDB_hg19_150331_TRgt6_lt101bp_gt95identity_merged | Tandem repeats with >6bp unit size and <101bp in length and >95% identity |
lowcmp_AllRepeats_gt200bp_gt95identity_merged | All perfect and imperfect tandem repeats >200bp in length |
lowcmp_Human_Full_Genome_TRDB_hg19_150331_TRgt6_51to200bp_gt95identity_merged | Tandem repeats with >6bp unit size and 51-200bp in length and >95% identity |
lowcmp_Human_Full_Genome_TRDB_hg19_150331_all_gt95identity_merged | All tandem repeats from TRDB with >95% identity |
lowcmp_SimpleRepeat_triTR_gt200 | Exact 3bp tandem repeats >200bp in length |
lowcmp_SimpleRepeat_triTR_51to200 | Exact 3bp tandem repeats 51-200bp in length |
lowcmp_Human_Full_Genome_TRDB_hg19_150331_TRlt7_lt51bp_gt95identity_merged | Tandem repeats with 1-6bp unit size and <51bp in length and >95% identity |
lowcmp_Human_Full_Genome_TRDB_hg19_150331_TRlt7_51to200bp_gt95identity_merged | Tandem repeats with 1-6bp unit size and 51-200bp in length and >95% identity |
lowcmp_AllRepeats_51to200bp_gt95identity_merged | All perfect and imperfect tandem repeats 51-200bp in length |
lowcmp_Human_Full_Genome_TRDB_hg19_150331 | All tandem repeats from TRDB |
lowcmp_SimpleRepeat_quadTR_51to200 | Exact 4bp tandem repeats 51-200bp in length |
lowcmp_AllRepeats_lt51bp_gt95identity_merged | All perfect and imperfect tandem repeats <51bp in length |
lowcmp_SimpleRepeat_homopolymer_6to10 | All homopolymers 6-10bp in length |
lowcmp_SimpleRepeat_diTR_gt200 | Exact 2bp tandem repeats >200bp in length |
lowcmp_SimpleRepeat_diTR_11to50 | Exact 2bp tandem repeats 11-50bp in length |
lowcmp_SimpleRepeat_diTR_51to200 | Exact 2bp tandem repeats 51-200bp in length |
lowcmp_Human_Full_Genome_TRDB_hg19_150331_TRgt6_gt200bp_gt95identity_merged | Tandem repeats with >6bp unit size and >200bp in length and >95% identity |
segdup | Segmental duplications in GRCh37 of all sizes, not including ALTs from GRCh37 |
segdupwithalt | Segmental duplications in GRCh37 >10kb, including ALTs from GRCh37 |
decoy | Regions in GRCh37 to which decoy sequences in hs37d5 map |
HG001compoundhet | Compound heterozygous regions in which there are 2 different variants when phased variants within 50bp are combined in HG001/NA12878 |
HG002compoundhet | Compound heterozygous regions in which there are 2 different variants when phased variants within 50bp are combined in HG002/NA24385 |
HG001complexvar | Complex variant regions in which there are 2 or more variants within 50bp in HG001/NA12878 |
HG002complexvar | Complex variant regions in which there are 2 or more variants within 50bp in HG002/NA24385 |
These dimensions help slice and dice the results according to all the different ways in which someone may want to look at them. The special value of '*' (for Subtype, Genotype, Subset) corresponds to summary statistics (i.e. as calculated across the whole genome in the confident regions, without further stratification).
We ran hap.py (and specifically the HAP-207 version, with the engine set to a GA4GH-specific version of vcfeval) to compare HG001 and HG002 submissions against the GiaB/NIST v3.2.2 HG001 and HG002 truth data respectively. Our executions excluded the entry labeled ccogle-snppet as it did not call variants in the whole genome. The entries labeled ghariani-varprowl, jpowers-varprowl and qzeng-custom contained few VCF lines which were deemed incompatible by this new comparison framework (which performs stricter checks). Upon closer inspection, this was due to incompatible REF columns (such as the strings "nan" or "AC-7GATAGAA") or non-diploid genotypes (such as 0/1/2, or even 0/1/2/3). These were a small fraction, so we decided to exclude these offending lines from this comparison.
We have made available on precisionFDA complete archives of all the input files (including both the original and adjusted files, for the entries which we had to remove offending VCF lines), and the results (including the annotated VCF as output by hap.py, and extended statistics in CSV format). These are recapitulated in a precisionFDA post.
We would like to thank the benchmarking group of the Global Alliance for Genomics and Health for their excellent collaboration throughout this precisionFDA effort.
| Entry | Type | Subtype | Subset | Genotype | F-score | Recall | Precision | Frac_NA | Truth TP | Truth FN | Query TP | Query FP | FP gt | % FP ma |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
85301-85350 / 86044 show all | ||||||||||||||
| gduggal-snapplat | SNP | tv | tech_badpromoters | * | 89.0511 | 84.7222 | 93.8462 | 77.3519 | 61 | 11 | 61 | 4 | 0 | 0.0000 |
| gduggal-snapplat | SNP | tv | tech_badpromoters | het | 87.8788 | 87.8788 | 87.8788 | 83.6634 | 29 | 4 | 29 | 4 | 0 | 0.0000 |
| gduggal-snapplat | SNP | tv | tech_badpromoters | hetalt | 0.0000 | 0.0000 | 0.0000 | 0 | 0 | 0 | 0 | 0 | ||
| gduggal-snapplat | SNP | tv | tech_badpromoters | homalt | 90.1408 | 82.0513 | 100.0000 | 62.3529 | 32 | 7 | 32 | 0 | 0 | |
| gduggal-snapfb | SNP | tv | lowcmp_Human_Full_Genome_TRDB_hg19_150331_TRlt7_51to200bp_gt95identity_merged | * | 15.1782 | 85.4545 | 8.3287 | 79.8084 | 141 | 24 | 151 | 1662 | 21 | 1.2635 |
| gduggal-snapfb | SNP | tv | lowcmp_Human_Full_Genome_TRDB_hg19_150331_TRlt7_51to200bp_gt95identity_merged | het | 13.3646 | 83.0645 | 7.2669 | 75.3956 | 103 | 21 | 113 | 1442 | 13 | 0.9015 |
| gduggal-snapfb | SNP | tv | lowcmp_Human_Full_Genome_TRDB_hg19_150331_TRlt7_51to200bp_gt95identity_merged | hetalt | 0.0000 | 0.0000 | 75.0000 | 0 | 0 | 0 | 37 | 0 | 0.0000 | |
| gduggal-snapfb | SNP | tv | lowcmp_Human_Full_Genome_TRDB_hg19_150331_TRlt7_51to200bp_gt95identity_merged | homalt | 29.0076 | 92.6829 | 17.1946 | 91.1987 | 38 | 3 | 38 | 183 | 8 | 4.3716 |
| gduggal-snapfb | SNP | tv | lowcmp_Human_Full_Genome_TRDB_hg19_150331_TRlt7_gt200bp_gt95identity_merged | * | 0.0000 | 100.0000 | 0 | 0 | 0 | 0 | 0 | |||
| gduggal-snapfb | SNP | tv | lowcmp_Human_Full_Genome_TRDB_hg19_150331_TRlt7_gt200bp_gt95identity_merged | het | 0.0000 | 100.0000 | 0 | 0 | 0 | 0 | 0 | |||
| gduggal-snapfb | SNP | tv | lowcmp_Human_Full_Genome_TRDB_hg19_150331_TRlt7_gt200bp_gt95identity_merged | hetalt | 0.0000 | 0.0000 | 0.0000 | 0 | 0 | 0 | 0 | 0 | ||
| gduggal-snapfb | SNP | tv | lowcmp_Human_Full_Genome_TRDB_hg19_150331_TRlt7_gt200bp_gt95identity_merged | homalt | 0.0000 | 100.0000 | 0 | 0 | 0 | 0 | 0 | |||
| gduggal-snapfb | SNP | tv | lowcmp_Human_Full_Genome_TRDB_hg19_150331_TRlt7_lt101bp_gt95identity_merged | * | 35.0458 | 95.2883 | 21.4714 | 85.0675 | 1355 | 67 | 1398 | 5113 | 85 | 1.6624 |
| gduggal-snapfb | SNP | tv | lowcmp_Human_Full_Genome_TRDB_hg19_150331_TRlt7_lt101bp_gt95identity_merged | het | 28.5443 | 93.7568 | 16.8348 | 81.4803 | 856 | 57 | 901 | 4451 | 48 | 1.0784 |
| gduggal-snapfb | SNP | tv | lowcmp_Human_Full_Genome_TRDB_hg19_150331_TRlt7_lt101bp_gt95identity_merged | hetalt | 2.8369 | 100.0000 | 1.4388 | 74.9550 | 2 | 0 | 2 | 137 | 3 | 2.1898 |
| gduggal-snapfb | SNP | tv | lowcmp_Human_Full_Genome_TRDB_hg19_150331_TRlt7_lt101bp_gt95identity_merged | homalt | 64.9197 | 98.0276 | 48.5294 | 92.7910 | 497 | 10 | 495 | 525 | 34 | 6.4762 |
| gduggal-snapfb | SNP | tv | lowcmp_Human_Full_Genome_TRDB_hg19_150331_TRlt7_lt51bp_gt95identity_merged | * | 41.5632 | 96.5217 | 26.4836 | 86.5727 | 1221 | 44 | 1254 | 3481 | 67 | 1.9247 |
| gduggal-snapfb | SNP | tv | lowcmp_Human_Full_Genome_TRDB_hg19_150331_TRlt7_lt51bp_gt95identity_merged | het | 33.9862 | 95.3401 | 20.6789 | 83.3507 | 757 | 37 | 792 | 3038 | 37 | 1.2179 |
| gduggal-snapfb | SNP | tv | lowcmp_Human_Full_Genome_TRDB_hg19_150331_TRlt7_lt51bp_gt95identity_merged | hetalt | 3.8462 | 100.0000 | 1.9608 | 75.9434 | 2 | 0 | 2 | 100 | 3 | 3.0000 |
| gduggal-snapfb | SNP | tv | lowcmp_Human_Full_Genome_TRDB_hg19_150331_TRlt7_lt51bp_gt95identity_merged | homalt | 72.4427 | 98.5075 | 57.2852 | 93.2156 | 462 | 7 | 460 | 343 | 27 | 7.8717 |
| gduggal-snapfb | SNP | tv | lowcmp_Human_Full_Genome_TRDB_hg19_150331_all_gt95identity_merged | * | 54.3229 | 97.9257 | 37.5868 | 83.5805 | 4060 | 86 | 4112 | 6828 | 99 | 1.4499 |
| gduggal-snapfb | SNP | tv | lowcmp_Human_Full_Genome_TRDB_hg19_150331_all_gt95identity_merged | het | 46.2642 | 97.2779 | 30.3489 | 80.4847 | 2573 | 72 | 2627 | 6029 | 58 | 0.9620 |
| gduggal-snapfb | SNP | tv | lowcmp_Human_Full_Genome_TRDB_hg19_150331_all_gt95identity_merged | hetalt | 3.6810 | 100.0000 | 1.8750 | 78.4657 | 3 | 0 | 3 | 157 | 3 | 1.9108 |
| gduggal-snapfb | SNP | tv | lowcmp_Human_Full_Genome_TRDB_hg19_150331_all_gt95identity_merged | homalt | 81.8789 | 99.0654 | 69.7740 | 90.1347 | 1484 | 14 | 1482 | 642 | 38 | 5.9190 |
| gduggal-snapfb | SNP | tv | lowcmp_Human_Full_Genome_TRDB_hg19_150331_all_merged | * | 73.1099 | 94.8597 | 59.4736 | 81.1920 | 13822 | 749 | 13965 | 9516 | 255 | 2.6797 |
| gduggal-snapfb | SNP | tv | lowcmp_Human_Full_Genome_TRDB_hg19_150331_all_merged | het | 66.2291 | 94.8867 | 50.8665 | 79.7967 | 8666 | 467 | 8806 | 8506 | 164 | 1.9281 |
| gduggal-snapfb | SNP | tv | lowcmp_Human_Full_Genome_TRDB_hg19_150331_all_merged | hetalt | 13.6364 | 100.0000 | 7.3171 | 82.8308 | 15 | 0 | 15 | 190 | 8 | 4.2105 |
| gduggal-snapfb | SNP | tv | lowcmp_Human_Full_Genome_TRDB_hg19_150331_all_merged | homalt | 90.3235 | 94.7999 | 86.2508 | 84.2900 | 5141 | 282 | 5144 | 820 | 83 | 10.1220 |
| gduggal-snapfb | SNP | tv | lowcmp_SimpleRepeat_diTR_11to50 | * | 73.6686 | 98.2908 | 58.9111 | 74.8889 | 4773 | 83 | 4826 | 3366 | 126 | 3.7433 |
| gduggal-snapfb | SNP | tv | lowcmp_SimpleRepeat_diTR_11to50 | het | 67.7466 | 98.0570 | 51.7501 | 74.5351 | 3028 | 60 | 3090 | 2881 | 75 | 2.6033 |
| gduggal-snapfb | SNP | tv | lowcmp_SimpleRepeat_diTR_11to50 | hetalt | 1.9417 | 100.0000 | 0.9804 | 62.5000 | 1 | 0 | 1 | 101 | 2 | 1.9802 |
| gduggal-snapfb | SNP | tv | lowcmp_SimpleRepeat_diTR_11to50 | homalt | 89.5049 | 98.6984 | 81.8782 | 76.1990 | 1744 | 23 | 1735 | 384 | 49 | 12.7604 |
| gduggal-snapfb | SNP | tv | lowcmp_SimpleRepeat_diTR_51to200 | * | 4.1580 | 76.9231 | 2.1368 | 79.9012 | 20 | 6 | 20 | 916 | 7 | 0.7642 |
| gduggal-snapfb | SNP | tv | lowcmp_SimpleRepeat_diTR_51to200 | het | 3.0730 | 70.5882 | 1.5707 | 75.7691 | 12 | 5 | 12 | 752 | 2 | 0.2660 |
| gduggal-snapfb | SNP | tv | lowcmp_SimpleRepeat_diTR_51to200 | hetalt | 0.0000 | 0.0000 | 64.7727 | 0 | 0 | 0 | 31 | 1 | 3.2258 | |
| gduggal-snapfb | SNP | tv | lowcmp_SimpleRepeat_diTR_51to200 | homalt | 10.6667 | 88.8889 | 5.6738 | 90.0424 | 8 | 1 | 8 | 133 | 4 | 3.0075 |
| gduggal-snapfb | SNP | tv | lowcmp_SimpleRepeat_diTR_gt200 | * | 0.0000 | 100.0000 | 0 | 0 | 0 | 0 | 0 | |||
| gduggal-snapfb | SNP | tv | lowcmp_SimpleRepeat_diTR_gt200 | het | 0.0000 | 100.0000 | 0 | 0 | 0 | 0 | 0 | |||
| gduggal-snapfb | SNP | tv | lowcmp_SimpleRepeat_diTR_gt200 | hetalt | 0.0000 | 0.0000 | 0.0000 | 0 | 0 | 0 | 0 | 0 | ||
| gduggal-snapfb | SNP | tv | lowcmp_SimpleRepeat_diTR_gt200 | homalt | 0.0000 | 100.0000 | 0 | 0 | 0 | 0 | 0 | |||
| gduggal-snapfb | SNP | tv | lowcmp_SimpleRepeat_homopolymer_6to10 | * | 99.2467 | 99.6334 | 98.8630 | 62.3274 | 10871 | 40 | 10869 | 125 | 24 | 19.2000 |
| gduggal-snapfb | SNP | tv | lowcmp_SimpleRepeat_homopolymer_6to10 | het | 99.1501 | 99.7008 | 98.6054 | 62.2072 | 6998 | 21 | 7000 | 99 | 15 | 15.1515 |
| gduggal-snapfb | SNP | tv | lowcmp_SimpleRepeat_homopolymer_6to10 | hetalt | 83.3333 | 100.0000 | 71.4286 | 70.8333 | 5 | 0 | 5 | 2 | 0 | 0.0000 |
| gduggal-snapfb | SNP | tv | lowcmp_SimpleRepeat_homopolymer_6to10 | homalt | 99.4469 | 99.5112 | 99.3827 | 62.5253 | 3868 | 19 | 3864 | 24 | 9 | 37.5000 |
| gduggal-snapfb | SNP | tv | lowcmp_SimpleRepeat_homopolymer_gt10 | * | 0.0000 | 100.0000 | 0 | 0 | 0 | 0 | 0 | |||
| gduggal-snapfb | SNP | tv | lowcmp_SimpleRepeat_homopolymer_gt10 | het | 0.0000 | 100.0000 | 0 | 0 | 0 | 0 | 0 | |||
| gduggal-snapfb | SNP | tv | lowcmp_SimpleRepeat_homopolymer_gt10 | hetalt | 0.0000 | 100.0000 | 0 | 0 | 0 | 0 | 0 | |||
| gduggal-snapfb | SNP | tv | lowcmp_SimpleRepeat_homopolymer_gt10 | homalt | 0.0000 | 100.0000 | 0 | 0 | 0 | 0 | 0 | |||
| gduggal-snapfb | SNP | tv | lowcmp_SimpleRepeat_quadTR_11to50 | * | 91.9883 | 99.2888 | 85.6879 | 49.6867 | 7399 | 53 | 7430 | 1241 | 43 | 3.4650 |
| gduggal-snapfb | SNP | tv | lowcmp_SimpleRepeat_quadTR_11to50 | het | 88.7788 | 99.0616 | 80.4299 | 50.4389 | 4645 | 44 | 4677 | 1138 | 26 | 2.2847 |
The interactive explorer includes an additional column, "% FP MA", indicating the percentage of the false positives which have matching non-ref alleles (i.e. FP.gt/QUERY.FP). For SNPs, this fraction varies greatly between pipelines, but for the majority it is less than 30%. Different callers would have the fewest FPs if these matching allele false positives were excluded from the FP counts, making clear that how performance metrics are defined can have a significant effect. For indels, this fraction varies as well, but is generally higher than 50%.
Using the interactive explorer you can also filter for different subsets, such as coding regions. Nonsurprisingly, there are very few FPs and FNs in coding regions for the accurate methods.
You can also see the fraction of calls that fall outside the confidence regions (reported as FRAC_NA). For SNPs, the fraction of calls outside GiaB high-confidence regions is ~10-25%, and for indels the fraction is higher than 50% for all methods. This is an important caveat to consider, since accuracy might be lower outside GiaB high-confidence regions.
Notably, most entries did much better at SNP accuracy measures than at indel accuracy measures. The majority of entries have high SNP f-score, recall, and precision -- above 99%. The respective numbers are lower for indels, and in fact indel recall is usually lower than indel precision. It is important to note, as mentioned in the truth data announcement, that the high-confidence calls have an increased FP, FN, and genotyping error rate for single base indels in homopolymers. This may be a contributing factor to the overall lower indel performance numbers.
Just like in the precisionFDA Consistency Challenge, it appears that pipelines are overall tuned for precision. Compared to the previous challenge, however, the input datasets had higher coverage, which may be a contributing factor to higher recall.
We've determined the winners by taking the highest value in each metric (F-score, Recall, Precision) per variant type (SNPs vs indels). Please note that the award for highest f-score has been called "highest performance", to reflect the nature of the score. For each combination of metric per variant type, we are also recognizing entries with high values of that metric, based on a cutoff at the first substantial drop in the percentages. These cutoffs are ultimately subjective, and not directly applicable to other regulatory contexts (which may require well-defined thresholds ahead of time).
These awards and recognitions are meant to encourage the community to participate in challenges and do not constitute an endorsement by the FDA.
We strongly urge the community to be cautious when interpreting these results. By their nature, high-confidence variant calls and regions tend to include a subset of variants and regions that are easier to characterize. Therefore, such benchmarking against the high-confidence truth data may in fact overestimate accuracy. Manual curation of sequence data in a genome browser for a subset of false positives and false negatives is essential for an accurate understanding of statistics like sensitivity and precision.
When interpreting results within stratification sub-categories, it may be useful to calculate confidence intervals around statistics like sensitivity because there may be very few examples of variants of some types in the benchmark calls in the stratification sub-category.
These results are based on the HG002 truth data. Pipelines may perform differently when confronted with other samples. For a more complete performance assessment of software pipelines, additional samples and experiments would be required.
We wanted to present an example of other ways in which this dataset can be analyzed, as a way to stimulate the community to conduct further experimentation with the precisionFDA Truth Challenge entries. Since the HG002 sample is male (in contrast to HG001, which is female), we decided to look at the performance of variation calling pipelines in chromosome X. It should be noted that chromosome X is not included in the confident regions of the GiaB truth data for HG002, so the comparisons have not evaluated that chromosome.
In XY male individuals, locations in chromosome X outside of the pseudoautosomal regions are expected to behave as haploid, and variant calling algorithms would ideally report them as haploid calls or as diploid homozygous calls. To measure that, we used bcftools stats -f .,PASS -r X:1-60001,X:2699521-154931044,X:155260561-155270560, which measures the count of heterozygous and homozygous SNPs in chromosome X outside of the PAR. We subsequently calculated the fraction of SNPs that are heterozygous, and generated the following table.
| Label | PctHet |
|---|---|
| dgrover-gatk | 0.00% |
| jli-custom | 0.00% |
| ltrigg-rtg1 | 0.00% |
| ltrigg-rtg2 | 0.00% |
| mlin-fermikit | 0.51% |
| ckim-isaac | 1.10% |
| raldana-dualsentieon | 1.66% |
| hfeng-pmm1 | 1.76% |
| hfeng-pmm3 | 1.97% |
| hfeng-pmm2 | 2.04% |
| ndellapenna-hhga | 2.21% |
| egarrison-hhga | 2.30% |
| bgallagher-sentieon | 2.34% |
| astatham-gatk | 2.36% |
| rpoplin-dv42 | 2.42% |
| ckim-dragen | 4.17% |
| cchapple-custom | 5.11% |
| ckim-gatk | 5.23% |
| ckim-vqsr | 5.23% |
| asubramanian-gatk | 6.00% |
| ciseli-custom | 6.10% |
| qzeng-custom | 6.23% |
| jlack-gatk | 6.32% |
| gduggal-bwafb | 7.89% |
| jpowers-varprowl | 8.52% |
| eyeh-varpipe | 10.62% |
| ghariani-varprowl | 10.94% |
| gduggal-snapfb | 11.32% |
| gduggal-bwavard | 13.09% |
| gduggal-bwaplat | 16.11% |
| gduggal-snapvard | 16.21% |
| gduggal-snapplat | 17.29% |
| anovak-vg | 31.01% |
Certain algorithms (such as the ones used in the ltrigg-rtg1 and ltrigg-rtg2 entries) can be gender-aware, and will not generate heterozygous calls when configured to run with a male option. Four entries in total had a zero heterozygosity fraction.
We want to thank those of you who participated in this challenge! As with our first challenge, by participating and putting your results and your thoughts out in the public, you fulfilled the first and most important goal of this challenge – to engage and start sharing data.
This challenge created a rich and interesting dataset that we hope people will study further. This dataset includes not only the submitted VCF files but also the complete set of comparison results (annotated VCFs and calculated statistics). It represents the essence of this challenge, and can hopefully be useful to inform future approaches, not only in terms of regulatory science but also for reference materials and benchmarking methodologies. We hope the GiaB group will be able to make use of it to improve reference calls (particularly around indels), and the GA4GH benchmarking group will be able to use it to improve the comparison methodology. We are excited to see how else the community will use this data set in the future.
In closing, we would like to leave you with several encouraging quotes from our collaborators and larger community, which will hopefully inspire all readers to actively engage with the precisionFDA community, or participate in one of our upcoming challenges:
“We hope that in the near future, thanks to more well-characterized truth sets, the genomics community can determine whether these more-refined pipelines are indeed beneficial for enabling Precision Data for Precision Medicine.”
“Thanks for your patience in being a guinea pig for our high confidence calls and benchmarking tools! This has been really useful for us already!”
“These challenges are potentially of as much value to those constructing truth sets as they are to the rest of the community, as they provide a means to look at a wide variety of call sets with respect to multiple truth sets and start to delve into potential biases in various truth sets and the methods used to construct them. This is an evolving process and it is great to see how GIAB and GA4GH are also moving things forward in this regard.”
“We applaud the effort by NIST and FDA to develop more truth sets that the accuracy of all algorithms can be benchmarked against. We look forward to more comprehensive truth sets and open challenges that can drive the development of better pipeline algorithms and enable precision data for precision medicine.”
“Thanks for all your help! Hopefully through this process we all learned and improved across multiple fronts, so it's already a huge success. I’m already thinking of ways some of these results could be used in publications about benchmarking.”